Modelling the conformational dynamics of poly-nucleotides
Biomolecules display diverse conformational changes depending on temperature and solvent conditions. Time scale of these changes vary in the range of picoseconds to seconds and define the biological function.
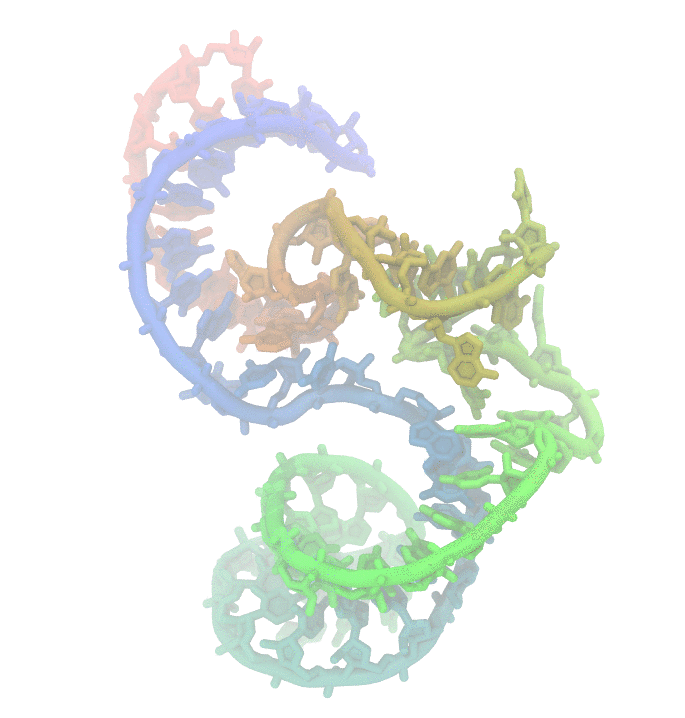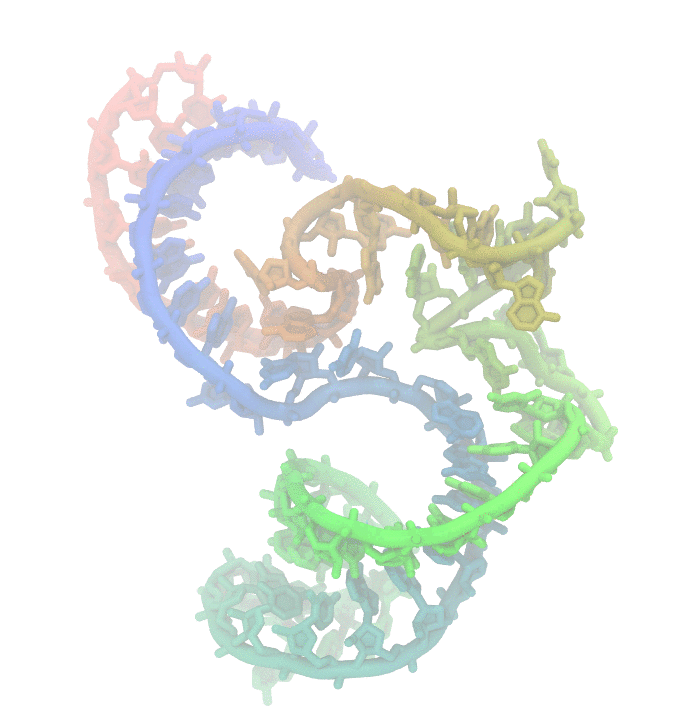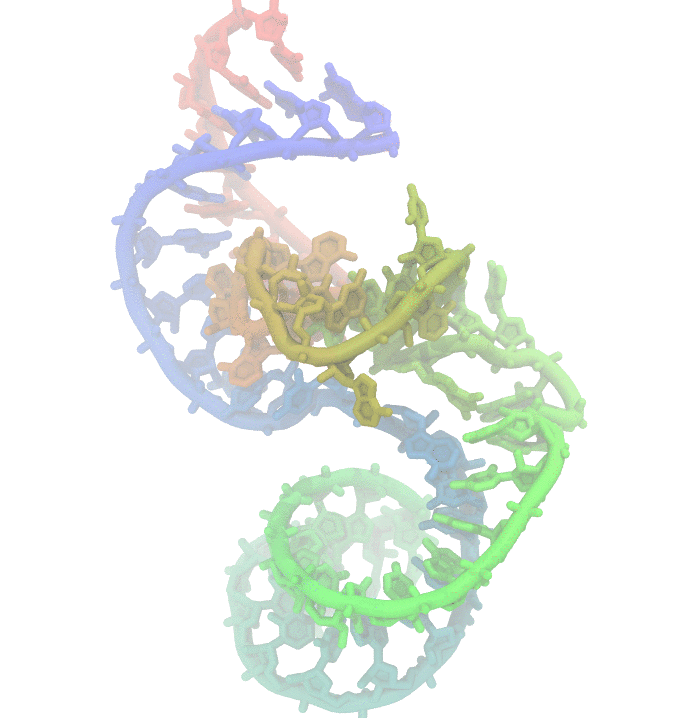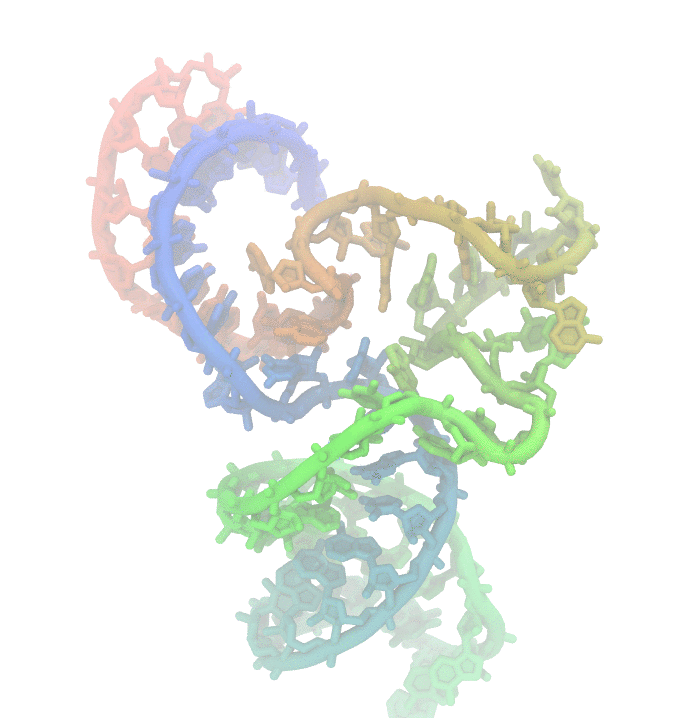
Fig 1. Conformational fluctuations of Hammerhead Ribozyme
Much effort has been invested to uncover the reasons behind structural changes and the effect of these changes on the function of biomolecules. We are particularly interested in nucleic acids. They display high structural changes that are highly depended on solvent conditions. The dynamic nature of poly-nucleotides makes experimental studies challanging. Using computer simulations such as molecular simulations is an alternative approach to study the biomolecular dynamics.
Despite the advancements in computational power, and the novel algorithms, it is yet a grand challange to reach the experimental time scales by conventional molecular dynamics simulations. Novel computational approaches need to be developed.
Here, we use and develop enhanced sampling methods to navigate the energy landscape. We use milestoning approach to connect the free energy basins explored to calculate the transition pathways and kinetics of conformational transitions.
To determine the slow conformational changes and to reduce the vast conformational space we employ machine learning methods.